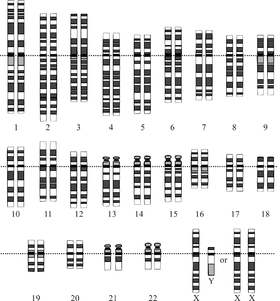
A graphical representation of the normal human
karyotype.
The human genome is the genome of Homo sapiens, which has 24 distinct chromosomes (22 autosomal + X + Y) with a total of approximately 3 billion DNA base pairs containing an estimated 20,000–25,000 genes. [1] The Human Genome Project has produced a reference sequence of the euchromatic human genome, which is used worldwide in biomedical sciences. The human genome had fewer genes than expected, with only about 1.5% coding for proteins, and the rest comprised by RNA genes, regulatory sequences, introns and controversially so-called junk DNA.[2]
Features
Chromosomes

The human genome is composed of 23 pairs of
chromosomes (46
in total), each of which contain hundreds of
genes separated by
intergenic regions. Intergenic regions may contain
regulatory sequences and non-coding DNA.
There are 24 distinct human chromosomes: 22 autosomal chromosomes, plus the sex-determining X and Y chromosomes. Chromosomes 1–22 are numbered roughly in order of decreasing size. Somatic cells usually have 23 chromosome pairs: one copy of chromosomes 1–22 from each parent, plus an X chromosome from the mother, and either an X or Y chromosome from the father, for a total of 46.
Genes
There are an estimated 20,000–25,000 human protein-coding genes [1]
Surprisingly, the number of human genes seems to be less than a factor of two greater than that of many much simpler organisms, such as the roundworm and the fruit fly. However, human cells make extensive use of alternative splicing to produce several different proteins from a single gene, and the human proteome is thought to be much larger than those of the aforementioned organisms.
Most human genes have multiple exons, and human introns are frequently much longer than the flanking exons.
Human genes are distributed unevenly across the chromosomes. Each chromosome contains various gene-rich and gene-poor regions, which seem to be correlated with chromosome bands and GC-content. The significance of these nonrandom patterns of gene density is not well understood. In addition to protein coding genes, the human genome contains thousands of RNA genes, including tRNA, ribosomal RNA, microRNA, and other non-coding RNA genes.
Regulatory sequences
The human genome has many different regulatory sequences which are crucial to controlling gene expression. These are typically short sequences that appear near or within genes. A systematic understanding of these regulatory sequences and how they together act as a gene regulatory network is only beginning to emerge from computational, high-throughput expression and comparative genomics studies.
Identification of regulatory sequences relies in part on evolutionary conservation. The evolutionary branch between the human and mouse, for example, occurred 70–90 million years ago.[3] So computer comparisons of gene sequences that identify conserved non-coding sequences will be an indication of their importance in duties such as gene regulation. [4]
Another comparative genomic approach to locating regulatory sequences in humans is the gene sequencing of the puffer fish. These vertebrates have essentially the same genes and regulatory gene sequences as humans, but with only one-eighth the "junk" DNA. The compact DNA sequence of the puffer fish makes it much easier to locate the regulatory genes.[5]
Other DNA
Protein-coding sequences (specifically, coding exons) comprise less than 1.5% of the human genome.[2] Aside from genes and known regulatory sequences, the human genome contains vast regions of DNA the function of which, if any, remains unknown. These regions in fact comprise the vast majority, by some estimates 97%, of the human genome size. Much of this is comprised of:
elements
However, there is also a large amount of sequence that does not fall under any known classification.
Much of this sequence may be an evolutionary artifact that serves no present-day purpose, and these regions are sometimes collectively referred to as "junk" DNA. There are, however, a variety of emerging indications that many sequences within are likely to function in ways that are not fully understood. Recent experiments using microarrays have revealed that a substantial fraction of non-genic DNA is in fact transcribed into RNA,[6] which leads to the possibility that the resulting transcripts may have some unknown function. Also, the evolutionary conservation across the mammalian genomes of much more sequence than can be explained by protein-coding regions indicates that many, and perhaps most, functional elements in the genome remain unknown.[7] The investigation of the vast quantity of sequence information in the human genome whose function remains unknown is currently a major avenue of scientific inquiry. [8]
Variation
Most studies of human genetic variation have focused on single nucleotide polymorphisms (SNPs), which are substitutions in individual bases along a chromosome. Most analyses estimate that SNPs occur on average somewhere between every 1 in 100 and 1 in 1,000 base pairs in the euchromatic human genome, although they do not occur at a uniform density. Thus follows the popular statement that "we are all, regardless of race, genetically 99.9% the same", [9] although this would be somewhat qualified by most geneticists. For example, a much larger fraction of the genome is now thought to be involved in copy number variation. [10] A large-scale collaborative effort to catalog SNP variations in the human genome is being undertaken by the International HapMap Project.
The genomic loci and length of certain types of small repetitive sequences are highly variable from person to person, which is the basis of DNA fingerprinting and DNA paternity testing technologies. The heterochromatic portions of the human genome, which total several hundred million base pairs, are also thought to be quite variable within the human population (they are so repetitive and so long that they cannot be accurately sequenced with current technology). These regions contain few genes, and it is unclear whether any significant phenotypic effect results from typical variation in repeats or heterochromatin.
Most gross genomic mutations in germ cells probably result in inviable embryos; however, a number of human diseases are related to large-scale genomic abnormalities. Down syndrome, Turner Syndrome, and a number of other diseases result from nondisjunction of entire chromosomes. Cancer cells frequently have aneuploidy of chromosomes and chromosome arms, although a cause and effect relationship between aneuploidy and cancer has not been established.
http://en.wikipedia.org/wiki/Human_genome



